
翻译生物学介绍
无细胞蛋白质合成(Cell-free protein synthesis,CFPS),也称为体外转录/翻译系统,是一种重要且通用的工具,可用于分子生物学家在基础和应用科学中阐明细胞途径和机制。无细胞蛋白质合成系统已成为高通量功能基因组学和蛋白组学中广泛应用且十分有效方法,相比于活细胞中的蛋白质表达,无细胞蛋白质合成系统能够提供更为显著的优势。
无细胞蛋白质表达的起源
19世纪末,Eduard Buchner首次提出了无细胞表达的基本原理,他通过在酵母提取物中将糖转化为二氧化碳和乙醇的方式阐述了这一概念(1)。
随后的几年里,为了回答“氨基酸究竟在蛋白质中起什么作用?”这一由来已久的问题,实验室开始采用这种技术用于蛋白质合成。1961年,科学家Marshall Nirenberg和Heinrich Matthaei在解决这一基本问题上实现了重大突破,成功地应用无细胞蛋白表达,在核苷酸三联体及其编码的氨基酸之间建立了联系。
他们使用基于大肠杆菌(E.coli)的体外翻译系统,可合成多肽聚苯丙氨酸。因此,他们确定了氨基酸苯丙氨酸及其对应密码子UUU之间的联系,从根本上发现了破解遗传密码的关键。这一突破性实验最终破解了所有剩余的氨基酸密码子,并为如今使用的各种翻译生物学系统奠定了基础(2)。
图1.上图描述了蛋白质-DNA结合。这种相互作用的其中一个例子是转录因子与DNA结合,可调节RNA[WH1] 合成。
翻译生物学的基本原理
尽管如今现有的系统范围广泛多样,但都属于两种无细胞表达系统的其中之一:Translation Systems(翻译系统)和Coupled Transcription and Translation(TnT®)Systems(转录翻译偶联系统)。尽量两种系统之间存在差异,但无细胞反应的基本原理是一致的。
无细胞表达开始于从培养的细胞中产生的粗提取物,这些细胞通常参与高速率的蛋白质合成,如未成熟的红细胞(网织红细胞)。这些粗提取物的内源性DNA和mRNA被去除,随后给细胞裂解产物补充进行翻译所需的大分子成分,例如核糖体、tRNA、氨酰tRNA合成酶和起始、延伸和终止因子。随后,通过添加合适的模板(DNA或mRNA)启动翻译程序,并在适当的温度下进行翻译。在Translation Systems中,利用纯化mRNA启动反应,而利用线性或质粒DNA模板启动的系统则统称为Coupled Transcription and Translation(TnT®)Systems(转录和翻译偶联(TnT®)系统)。
为确保翻译高效进行,每种提取物均需要额外补充氨基酸、能量来源(ATP、GTP)、能量再生系统和盐类(例如Mg2+、K+)。一般来说,磷酸肌酸和肌酸磷酸激酶是真核生物系统中的能量再生系统,而原核生物系统通常需要补充磷酸烯醇丙酮酸盐和丙酮酸激酶。此外,转录和翻译偶联(TnT®)系统通常也提供噬菌体RNA聚合酶(T3、T7或SP6),可从外源性DNA模板中转录mRNA,允许T3、T7或SP6启动子下游克隆基因的表达。
细胞提取物和系统选择
对于为您选择合适的无细胞蛋白表达系统,需要考虑很多因素,包括您将使用的模板类型、您期望获得的蛋白质产率以及预期的下游应用。
迄今为止,最常用的商业化体外翻译系统包括大肠杆菌、小麦胚芽、兔网织红细胞或昆虫细胞提取物。由于每种细胞的表现和作用方式不同,因此其衍生的提取物也各不相同,每种都具有各自的优势和缺点,下面将进行简要介绍(1;3)。
由于以下几点原因,原核生物大肠杆菌系统目前是最受欢迎的蛋白质表达系统。大肠杆菌在低成本培养基中容易大量发酵,并且在高压均质机中很容易破裂,因此大肠杆菌提取物的制备操作简单,价格低廉。一般来说,基于大肠杆菌的系统蛋白质产率可达到最高,大肠杆菌系统的总反应成本最低。大肠杆菌能够激活提取物中的代谢反应,进而为高水平的蛋白质合成提供能量,从而减少对磷酸烯醇丙酮酸盐等更昂贵的能量代谢底物的需要。
目前,小麦胚芽提取物(WGE)、兔网织红细胞裂解物(RRL)和昆虫细胞提取物(ICE)系统是应用最广泛的真核生物系统。这些系统在产成更复杂的蛋白质方面更具优势,也可完成大肠杆菌中无法完成的翻译后修饰。然而,这些真核生物系统涉及的提取制备程序通常更为复杂费力,可能会导致成本增加。相比于大肠杆菌系统,真核生物系统在批量反应中也更倾向于导致较低的蛋白质产率。
小麦胚芽和兔网织红细胞裂解物的无细胞提取物支持各种病毒、原核生物和真核生物mRNA的体外翻译。这些RNA驱动的系统广泛用于识别mRNA种类并对其产物进行表征。从感兴趣的DNA开始,利用RiboMAX™ Large Scale RNA Production Systems(目录号:P1280、P1300)和T7 RiboMAX™ Express Large Scale RNA Production System(目录号:P1320)产生用于翻译的体外转录物(5~80µg/ml)。
大肠杆菌S30提取物(ECE):
优势:提取物制备操作简单,性价比高。ECE系统能够折叠复杂蛋白质,始终具有较高的蛋白质合成率,以及由此产生的较高的蛋白质合成产率。此外,能量来源成本低廉,当前的方法容易掌握,同时具备完善的工具可进行基因修饰。
缺点:可选择的翻译后修饰的数量有限,无可用于合成整合性膜蛋白的内源性膜结构。
小麦胚芽提取物(WGE):
优势:利用WGE可重复实现真核蛋白的广谱表达。该系统的产量较高,可翻译产生高产率的复杂蛋白。该系统是一种先进的高通量蛋白组学方法。
缺点:裂解产物的制备价格昂贵,费时费力。翻译后修饰可能受限,无用于合成整合性膜蛋白的内源性膜结构,相比于原核生物系统,WGE的蛋白产率较低。
兔网织红细胞裂解物(RRL):
优势:细胞容易破裂,提取物制备过程耗时较短。RRL系统行之有效,系统完善。该系统适用于哺乳动物系统真核特异性修饰,蛋白质产率适中/较低。
缺点:蛋白质产率较低。仅能够通过补充外源性微粒体膜进行翻译后修饰。
昆虫细胞提取物(ICE):
优势:细胞容易破裂,提取物制备过程耗时较短。可使用该提取物实现多个真核特异性翻译后修饰,包括糖基化、二硫键形成、脂质化和信号肽切割磷酸化。也可使用内源性微粒体,而且已成功使用该提取物进行膜蛋白的直接合成和整合。
缺点:昆虫细胞提取物的培养成本较高。
准备好开始寻找你的表达系统了吗?探索我们的蛋白表达产品组合,为您的无细胞蛋白表达需求找到正确的解决方案。
无细胞蛋白表达系统的优势
相比于基于细胞的蛋白质表达,无细胞蛋白表达系统具有多个明显的优势,包括提高了功能性和可溶性全长蛋白的总产率,并且节省了大量时间。一般来说,体内蛋白质合成方法最少需要几天,最长需要几周时间才能完成(4)。体外翻译反应可仅在几小时内完成(包括制备提取物所用的时间),为表型(被表达的蛋白的功能)和基因型建立联系提供了最快的方式,这便与体内方法形成了鲜明对比。
使用无细胞方法时,蛋白质合成也是灵活多样的,因为可以使用多种方式进行。无细胞翻译系统利用mRNA作模板,而在转录/翻译偶联系统中,质粒DNA或线性PCR片段可作为DNA模板。
CFPS的另一优势:其是一种开放性反应系统。由于无细胞蛋白表达不受细胞壁的限制,因此有充分的机会可直接操控化学环境,可添加外源成分或分子,创造更有利于蛋白质折叠和活动的环境(5)。CFPS系统可主动监测,迅速取样和筛选,不需要基因克隆步骤(3)。
由于没有细胞障碍限制翻译控制,CFPS系统可作为一种较为理想的方法,用于合成传统方式难以表达和进行后续分析的蛋白质。在无内在细胞代谢或生化途径相冲突的情况下,膜蛋白、病毒蛋白、毒性蛋白以及通过细胞内过程进行快速蛋白水解降解的蛋白更容易表达。因此,可缓解对于生产蛋白的潜在毒性的担忧。

图2.已开发大量大规模蛋白质合成方法,将无细胞系统产率提高到制备规模。
从核酸可编程蛋白阵列(NAPPA)到使用展示技术的酶工程技术,各种无细胞蛋白表达系统可适用于广泛的蛋白质表征应用。无细胞方法还可通过修饰后带电荷的tRNA或氨基酸,利用荧光、生物素、放射性或重原子对特异性蛋白质进行标记。
真核生物无细胞蛋白表达
真核生物的无细胞表达系统(RRL、小麦胚芽或昆虫细胞提取物)是可通过mRNA启动的翻译系统,或是通过补充最优噬菌体RNA聚合酶(T7、SP6或T3)并以含有T7、SP6或T3启动子的质粒DNA或PCR DNA启动的转录/翻译偶联(TnT®)系统。真核生物无细胞偶联系统可将原核噬菌体RNA聚合酶与真核提取物相结合,并利用外源DNA或PCR生成的模板,与噬菌体启动子共同进行体外蛋白质合成(图3)。
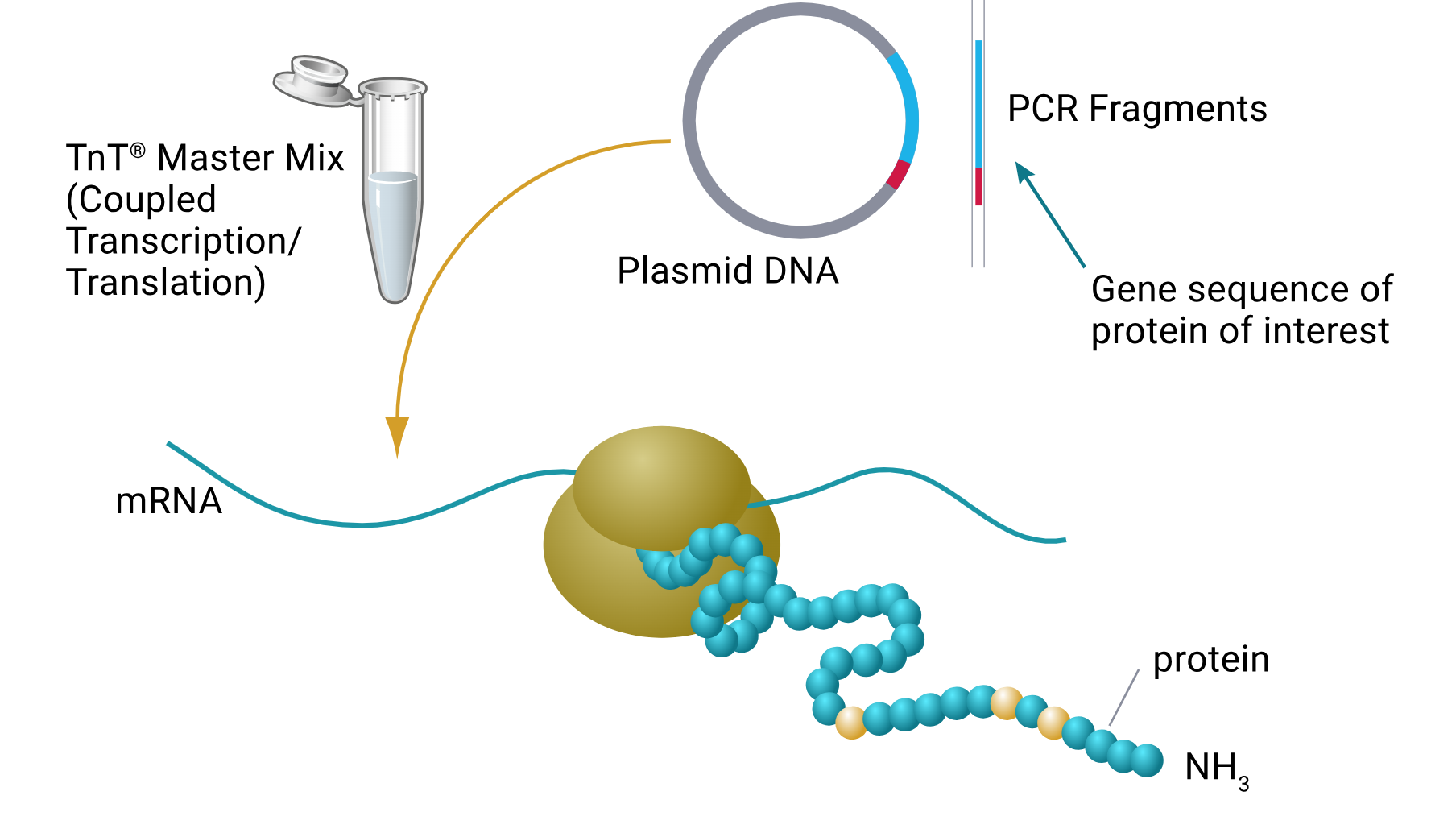
图3.利用TNT®系统的无细胞表达
无细胞转录和/或翻译系统提供了相当大的实用价值,尤其是在功能蛋白组学方面。特别是近年来,高产率表达系统的发展扩大了其应用范围(6)。
无细胞翻译系统
Rabbit Reticulocyte Lysate Translation Systems(Nuclease-treated and Untreated)和Wheat Germ Extract System可用于mRNA翻译。Rabbit Reticulocyte Lysate, Nuclease-Treated(目录号:L4960)可通过添加多种补充物进行优化,用于mRNA翻译。其中包含血红素,可防止血红素调节的eIF-2a激酶(HRI)的活化;能量再生系统包括预先测试的磷酸肌酸激酶和磷酸肌酸;小牛肝tRNA,可平衡可接受的tRNA的群体,从而优化密码子的使用,并扩大可有效翻译的mRNA范围。此外,裂解物经过微球菌核酸酶处理,消除了内源mRNA,因此,减小了本底的翻译。相比于Rabbit Reticulocyte Lysate, Nuclease-Treated,Flexi®Rabbit Reticulocyte Lysate System(目录号:L4540)提供了更加灵活的反应条件,通过对翻译反应在广泛的参数范围进行优化(包括Mg2+和K+浓度),并提供了添加DTT的选择。
相比于经处理的RRL,Rabbit Reticulocyte Lysate, Untreated(目录号:L4151)包含蛋白质合成需要的细胞成分(tRNA、核糖体、氨基酸、起始、延伸和终止因子),但未经过微球菌核酸酶处理。不建议使用Untreated Rabbit Reticulocyte Lysate进行特异性mRNA的体外翻译。
Wheat Germ Extract(目录号:L4380)包含蛋白质合成需要的细胞成分(tRNA、核糖体、起始、伸长和终止因子)。该提取物通过添加能量生成系统(包括磷酸肌酸和磷酸肌酸激酶)、亚腈胺(用于促进氨基酸链的延伸效率,从而克服过早终止)以及醋酸镁(推荐浓度是用于大多数mRNA翻译的浓度),进行进一步优化。只有添加外源氨基酸(包括相应标记的氨基酸)和mRNA才能够激活翻译程序。为进一步优化,可添加醋酸钾,用于促进多种mRNA的翻译。
无细胞转录/翻译系统
转录/翻译偶联系统通过将转录/翻译偶联至单管系统中,为研究人员提供了真核生物体外转录和翻译的选择方案,节省了时间。标准的RRL或WGE翻译(7)使用体外合成的RNA(8)。RNA被用作翻译模板。偶联系统(如TnT®系统)通过将转录需要的试剂直接添加至翻译混合物中来减少这些步骤。
在大多数情况下,相比于使用RNA模板的标准体外Rabbit Reticulocyte Lysate或Wheat Germ Extract翻译系统,TnT®System在1至2小时的反应中可显著提高蛋白质产量(两倍至六倍)。此外,TnT®Lysates可与微粒体膜一同用于研究蛋白质加工。
微粒体囊泡用于研究蛋白的共翻译和初始翻译后加工(9;10;11)。在这些微粒体膜存在的情况下,可通过相应的mRNA的体外翻译,对包括信号肽切割(12)、膜插入(13)、易位和核心糖基化(14)等在内的加工事件进行检测。加工和糖基化事件也可通过转录/翻译相应的DNA来研究。
Rabbit Reticulocyte Lysate系统的替代品包括小麦胚芽系统和使用昆虫细胞系提取物的系统,例如常用的Spodoptera frugiperdaSf21细胞系(15),该细胞系是TnT®T7 Insect Cell Extract Protein Expression System(目录号:L1101、L1102)的组分。相比于其他无细胞裂解产物,麦胚提取物无细胞蛋白合成系统提供了独特的优势,包括室温培养、高通量筛选、灵活添加辅助组分、细胞毒性蛋白的表达及蛋白折叠和功能的筛选(16;17)。
TnT®Quick Coupled Transcription/Translation System
TnT®Quick Coupled Transcription/Translation Systems通过将所有的反应组分(RNA聚合酶、核苷酸、盐类以及RNasin®Ribonuclease Inhibitor)连同网织红细胞裂解液一起包含在一个TnT®Quick Master Mix(TnT®快速混合母液)中,从而简化了转录/翻译的步骤(图4)。
该Master Mix的成分已经过仔细调整,可使大多数基因结构的表达和保真度最大化。如有需要,可使用醋酸镁和氯化钾,优化TnT®Quick Systems的体外翻译反应。
将RNasin®Ribonuclease Inhibitor直接加入Master Mix中,可防止引入使用某些小规模制备方案制备的DNA溶液中携带的RNases时产生的潜在危害。
TnT®Quick System有两种配置,分别对克隆在T7(目录号:L1170、L1171)或SP6(目录号:L2080、L2081)RNA聚合酶启动子下游的基因进行转录和翻译。有关该系统详细的方案和背景信息,请参看技术手册#TM045(Technical Manual #TM045)。
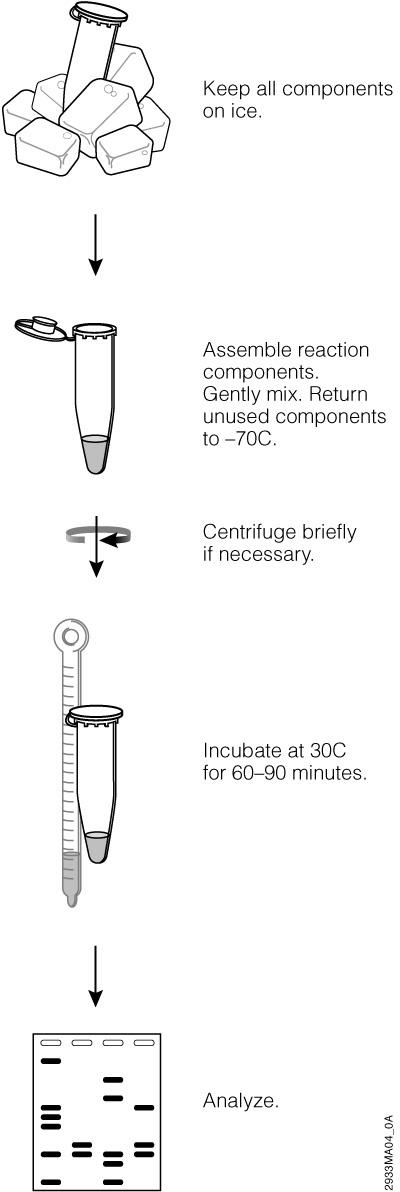
图4.描述TNT®系统方案的流程图。
The TNT®Quick Coupled Transcription/Translation Systems
The TNT®Quick Coupled Transcription/Translation Systems为真核细胞无蛋白表达提供方便的单管、偶联转录/翻译体系。
原核生物无细胞蛋白表达
大肠杆菌S30提取物系统
一般来说,大肠杆菌S30组分用于原核表达。尽管不应仅通过靶蛋白的来源确定系统的选择,但可根据蛋白的生物特性和下游应用的要求进行选择。大肠杆菌系统的产率优于真核生物系统,通常可达几mg/mL,主要取决于蛋白质和反应形式。
S30 T7 High-Yield Protein Expression System(S30 T7高产率蛋白表达系统)(目录号:L1110、L1115)是一种大肠杆菌提取物无细胞蛋白合成系统。通过提供含有用于转录的T7 RNA聚合酶和所有翻译需要的成分的提取物,简化了含有T7启动子的质粒中克隆的DNA序列的转录和翻译过程。该系统可使用包含感兴趣的序列、T7启动子和核糖体结合位点(RBS)的载体,在一小时内生成高水平的重组蛋白(每毫升反应高达数百微克重组蛋白)。
E.coli S30Extract Systems(大肠杆菌提取物系统)中的S30提取物可通过经过调整的Zubay所述的方法进行制备(18;19;20)。对于线性模板表达(目录号:L1030),提取物是从ompT内蛋白酶和离子蛋白酶活性缺乏的大肠杆菌B菌株中制备而来。这使得表达的蛋白质具有更高的稳定性,如果在体内表达,这些蛋白质则会被蛋白酶降解(21;22)。如果使用环状DNA模板,则E.coliS30Extract Systems(目录号:L1020)可生成更高表达水平的蛋白质,而这些蛋白质由于宿主编码阻遏物的作用在体内的表达水平一般较低(23)。优化的S30 Premix Plus提供了表达高水平重组蛋白所需的所有其他组分。
E.coliS30 Extract Systems中表达的蛋白质可用于多种功能性转录和翻译研究。E.coli S30 Extract Systems最常用于合成少量放射性标记蛋白,可用作蛋白质纯化中的示踪剂,也可用于将非天然氨基酸添加至蛋白质中进行结构研究(24)。
无细胞蛋白表达系统的应用
随着我们对无细胞表达系统及其功能认识的不断加深,研究人员已经能够利用这些系统的各种优势,开发新型蛋白质技术。下面我们将探讨这些独特的应用。
功能基因组和蛋白质组分析
无细胞蛋白质合成提供了一种基础简单的工具,帮助识别蛋白质和分子的相互作用,例如蛋白质-蛋白质、蛋白质-核酸以及蛋白质-配体。为了表征这些相互作用,对其中一种研究对象(核苷酸/配体/蛋白质等)进行标记,随后与其他研究对象一同放入CFPS进行培养。之后,利用如免疫沉淀法等技术分离所产生的复合物,或者利用电泳迁移率变化检测(EMSA)直接检测复合物,其中,相比于未结合的复合物,蛋白质相互作用复合物的电泳速度减慢(5)。
无细胞蛋白表达系统对于制造蛋白质阵列同样是十分重要的工具。蛋白质微阵列是固相的配体结合分析系统,可用于追踪蛋白质活性和相互作用,并用于检测高通量形式中的蛋白质功能。传统的基于细胞的蛋白质芯片的生成是一个劳动密集型的过程,需要表达和纯化每个单独的蛋白质进行排列。另一个缺点是固定化蛋白质的长期功能稳定性通常受限。使用CFPS可通过在原位(芯片上)直接并行合成多种蛋白质来克服这些挑战。
多个不同种类的蛋白质微阵列可用于当前研究,包括PISA(蛋白质原位阵列)、NAPPA(核酸可编程蛋白质阵列)和DAPA(DNA阵列到蛋白质阵列)。
图5.体外无细胞系统中生成的蛋白质展现了各种蛋白质功能或活性,这取决于正确合成、折叠和辅助因子结合等必须的因素。上图中,只要存在必要的辅助因子ATP,蛋白质便会显示发光信号。
PISA
PISA方法是首个为人熟知的无细胞原位蛋白质微阵列技术,可通过溶液中的无细胞表达,快速从DNA片段中直接生成标记蛋白。编码目的蛋白的单个DNA构建体包含T7启动子、用于翻译启动和终止的序列以及N端或C端标记序列。利用高保真TAQ聚合酶通过PCR或RT-PCR生成DNA构建体,随后,用无细胞转录和翻译偶联系统表达所需标记蛋白(25)。合成后,通过载玻片表面包被的标记捕获涂层,同时捕获蛋白质,并将其固定在原位。
NAPPA
NAPPA是一种标记技术,其中,DNA模板可生物素化,并固定在包被亲和素和抗GST抗体(可用作蛋白质捕获试剂)的载玻片上(27)。随后,该阵列可通过使用兔网织红细胞裂解物或类似CFPS系统,用于在原位表达靶蛋白。翻译后,固定化抗体可在每个位点捕获靶蛋白,从而形成蛋白质阵列,其中每个蛋白质与其相应的表达质粒共定位(28)。
NAPPA方法在描述疾病生物标志物和研究自身免疫病方面展现了其重要性。NAPPA生成的微阵列已用于帮助识别对结核分枝杆菌全蛋白质组的新抗体应答,准确指出了8种具有结核病生物标志物价值的蛋白质(29)。NAPPA还用于描述自身免疫性风湿性疾病(强直性脊柱炎)患者的自身抗体反应特征(30),还可用于表征青少年特发性关节炎患者的循环和滑膜抗体(31),以及用于检测可结合乳腺癌肿瘤抗原的抗体(32)。
DAPA
DNA阵列至蛋白质阵列(DAPA)方法的开发,作为一种通过从单个DNA阵列模板中按需打印生成蛋白质阵列的手段。在该方法中,含有可编码一系列靶蛋白的大量固定PCR扩增片段的载玻片与第二张预先包被蛋白质标记捕获试剂的载玻片进行面对面组装。随后,将包含无细胞裂解物的渗透膜放置在两张载玻片之间,从而实现转录和翻译偶联。蛋白质合成来源于固定的DNA位点,合成蛋白质通过膜扩散,并被固定在捕获载玻片表面,从而形成蛋白质阵列(33)。
表达困难的蛋白质的表达
无细胞蛋白质表达系统提供了一种生产通常难以表达的功能性蛋白质的方法,例如膜蛋白、毒性蛋白和病毒蛋白。使用偶联CFPS系统时,在适当的氧化和折叠条件下,已成功且完整地组装了复杂的异四聚体和多重二硫桥联的IgG分子(34)。各种无细胞系统也成功地用于体外合成膜蛋白,包括G蛋白偶联受体、表皮生长因子受体和ATP合成酶(35)。
体外翻译系统也已成功用于表达病毒蛋白、病毒样颗粒(VLPs)和疫苗抗原。在一项研究中,要体外合成124个感兴趣的恶性疟原虫(P. falciparum)目的基因作为潜在的疟疾候选疫苗,结果无需密码子优化就成功地以可溶性形式合成了75%的产物。无细胞大肠杆菌提取物系统已用于合成VLPs和疫苗,包括抗流感VLPs、B细胞淋巴瘤疫苗和抗乙型肝炎VLPs(35)。
体外系统有效地生成了传染性脑心肌炎病毒(EMCV),并且在补充犬微粒体膜时,已正确翻译并加工了丙型肝炎病毒RNA全部的开放阅读框(5)。这些研究表明,无细胞表达系统具有巨大的潜力,不仅可作为重要的工具用于了解病毒复制的机制,还可作为一种疫苗发现方法和前瞻性平台,用于大规模生产疫苗(5;35)。
蛋白质进化和酶工程技术
蛋白质是复杂、形式多样的分子,在细胞中提供多种功能。蛋白质定向进化是一种功能基因变体选择和基因多样化之间交替的循环过程,可作为改善某种蛋白质功能的工具,例如催化活性、结合亲和力和热稳定性(4;36)。在过去的三十年中,利用体外翻译系统开发了多种独特的蛋白质定向进化技术,包括核糖体展示、mRNA展示以及体外区室化。相比于细胞方法,这些体外技术提供了多种优势,包括遗传信息(基因型)及其编码的蛋白质(表型)的有效连接,以及更大范围的文库大小。
核糖体展示
核糖体展示方法可从多肽DNA序列文库中分离单个蛋白质。在体外翻译过程中,可通过连接新生多肽及其对应的编码mRNA建立蛋白质-核糖体-mRNA复合物(protein-ribosome-mRNA,PRM)(1)。
随后,可通过固定化配体的亲和力结合捕获目的PRM,然后从中重获mRNA,并将其逆转录成cDNA,根据需要进行突变。这一过程也可以只是重复进行而不突变回收的cDNA,作为从大量群体中富集目标蛋白的手段(5)。
核糖体展示技术(图6)是一种十分受欢迎的方法,可用于抗体、以及转录因子、受体、蛋白酶、酶、新肽、标签序列、配体结合域和基序等的体外选择和进化(5)。核糖体展示也可用于蛋白组学应用,从而鉴定出一段能够提高翻译效率的5'-非翻译区序列(37),以及用于作为潜在疫苗候选物的金黄色葡萄球菌cDNA文库抗原蛋白的综合测定(38)。
图6.上图描述了抗体-核糖体-mRNA(ARM)复合物,可用作体外真核生物方法中展示选择颗粒,用于选择抗体结合位点。ARM的重要特征是可保留目的肽与编码目的肽的基因之间的联系。
mRNA展示
在mRNA展示技术中,可编码目的多肽的大型DNA文库转录为mRNA。mRNA的3’端可连接至携带衔接分子(一般为嘌呤霉素)的短的单链DNA寡核苷酸上。利用无细胞蛋白质合成翻译修饰后的mRNA产物;嘌呤霉素进入核糖体内,与新生多肽链之间形成肽键。随后,通过逆转录生成cDNA,稳定核酸组分,并促进选择后遗传信息的回收。随后,可通过PCR扩增该cDNA,用于进一步研究或其他用途(39)。
该mRNA展示技术已成功用于从人工随机序列库中定向进化连接酶和ATP结合结构域(40;41。mRNA展示已用于获得基于III型支架纤维连接蛋白的抗原结合蛋白质,从而生成可结合肿瘤坏死因子(TNF-alpha)的高亲和力分子(42)并识别特异性内源性磷酸化IkBalpha(43)。
体外区室化
体外区室化(IVC)是一种定向进化方法,其目的旨在将较大的反应分隔为多个微观区室,可将DNA及其相关的mRNA和蛋白质封装在油包水乳胶微滴中或微珠表面。每个液滴包含一个基因,除此之外,还需要所有必要的分子组分,随后将其进行转录和翻译。表达所需活性的蛋白质可将底物转化为与基因保持联系的产物,这是因为它们均完全受限于微滴。与产物相连的基因可选择性富集、扩增和表征,或再与底物相连,并通过区室化进行其他选择。IVC方法已成功用于多个酶工程技术,包括甲基转移酶、聚合酶和限制性内切酶(5;44)。
筛选
将无细胞表达系统纳入高通量筛选应用中,同时满足蛋白质和多肽的原位和按需表达。这些无细胞表达高通量筛选应用大致分为芯片技术和体外展示,以及蛋白质进化和酶工程技术章节讨论过的一些选择方法。
在体外展示选择技术中,表达的复合物可直接进行筛选,并检测其活性,相关基因序列可用于连续富集。芯片选择技术将表达的蛋白质固定在经处理的表面,未使用其来源的DNA或RNA(45)。
当前存在多种高通量筛选方法,所有这些方法都能够简化传统筛选过程。这些筛选方法包括微量滴定板、数字成像、荧光活化细胞分选(fluorescence-activated cell sorting,FACS)、细胞表面显示、IVTC和共振能量转移(46)。
体外翻译后进行筛选已广泛用于医疗应用,使其开发了蛋白截断实验(Protein Truncated Test,PTT)(图7),其中使用了翻译-终止突变造成的遗传疾病的开放阅读框作为诊断(5)。无细胞蛋白质表达系统还可筛选毒性试剂(47),并识别翻译抑制剂作为潜在药物(48)。通过体外蛋白质表达筛选和生成潜在疫苗候选物可识别某些有效疫苗,这些疫苗能够保护小鼠免受肿瘤侵袭,其疗效与哺乳动物细胞中产生的相同(49)。
图7.该图描绘了使用蛋白质截断实验生成的截短蛋白(右)与正常长度蛋白质(左)的对比。体外进行该实验确定了基因突变是否导致缩短了翻译产物,从而导致生成癌细胞。
蛋白质标记与检测
对大多数应用,例如蛋白质:蛋白质相互作用和蛋白质:核酸相互作用研究来说,使用无细胞系统表达的蛋白质进行检测是必要的。在传统技术中,将放射性[35S]甲硫氨酸添加至无细胞表达反应中,甲硫氨酸可结合至表达的蛋白质中,利用放射自显影术进行检测。由于成本高昂、法规限制、放射性暴露以及废物处理问题,许多研究人员逐渐放弃了放射性技术。尽管传统的蛋白质印迹分析为研究人员提供了非放射性方法进行检测,但如果操作不当,可能会产生高背景。然而,例如FluoroTect™ GreenLysin vitro Translation Labeling System(目录号:L5001)和Transcend™ Non-Radioactive Translation Detection System(目录号:L5070、L5080)这些检测方法,使得蛋白质印迹检测更加灵敏,背景更低(50)。
FluoroTect™ System采用了带有赖氨酸的tRNA,在e位上用BODIPY®-FL荧光基团进行标记。体外翻译期间,可将这些荧光标记的赖氨酸残基掺入合成的蛋白质中。翻译过程中,Transcend™ System依赖于将生物素化赖氨酸残基掺入新生蛋白质中。该生物素化赖氨酸以预加电荷的e-标记生物素化赖氨酸:tRNA复合物(Transcend™ tRNA)而不是游离氨基酸的形式添加至转录/翻译反应中。SDS聚丙烯酰胺凝胶电泳(SDS-PAGE)和电印迹后,可以通过结合Streptavidin-AP或Streptavidin-HRP,然后分别通过比色或化学发光检测方法来观察生物素化蛋白质。一般来说,这些方法可检测0.5~5ng的蛋白质,其灵敏度与[35S]甲硫氨酸掺入和放射自显影检测的灵敏度相同。
Transcend™ Non-Radioactive Translation Detection Systems
Transcend™ Non-Radioactive Translation Detection Systems(Transcend™非放射性翻译检测系统)可对体外合成的蛋白质进行非放射性检测。使用该系统,可在翻译期间将生物素化赖氨酸残基掺入新生蛋白质中,消除利用[35S]甲硫氨酸或其他放射性氨基酸标记的需要。该生物素化赖氨酸以预加电荷的e-标记生物素化赖氨酸-tRNA复合物(Transcend™tRNA)而不是游离氨基酸的形式添加至转录/翻译反应中。SDS-PAGE和电印迹后,通过结合链霉亲和素碱性磷酸酶(Streptavidin-AP)或链霉亲和素辣根过氧化物酶(Streptavidin-HRP),然后通过比色或化学发光检测方法来观察生物素化的蛋白质。一般来说,可在凝胶电泳后3~4小时内检测到0.5~5ng的蛋白质。其灵敏度与凝胶电泳后6~12小时内的[35S]甲硫氨酸掺入和放射自显影检测的灵敏度相同。更多详细方案和背景信息请参见技术手册#TB182。
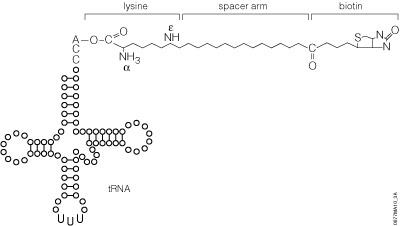
图8.Transcend™ tRNA结构示意图。
使用Transcend™ tRNA有多个优势:
- 无需进行放射性同位素处理或储存。
- 生物素标记可检测(0.5~5ng的灵敏度)。
- 不论是作为Transcend™ tRNA试剂,还是合成的蛋白中的标记,生物素标签可在12个月内稳定。与[35S]标记的蛋白质不同(其标记随时间消退),无需定期重新合成生物素标记的蛋白质。
- 无论标记的强度或凝胶上的负载量多少,标记的蛋白质均可检测到清晰的条带,因此,可检测表达不佳的基因产物。
- 利用比色或化学发光检测方法可快速观察到结果。
该系统中提供的预带电荷的大肠杆菌赖氨酸tRNA已使用经过修改的Johnson等人(1976)开发的方法在e-氨基酸处进行化学生物素化标记。生物素部分通过间隔臂与赖氨酸相结合,这极大地促进了亲和素/链霉亲和素试剂的检测(图8)。产生的生物素化赖氨酸tRNA分子(Transcend™ tRNA)可用于真核生物或原核生物体外翻译系统中,例如TNT®Coupled Transcription/Translation Systems、Rabbit Reticulocyte Lysate、Wheat Germ Extract或E. coliS30 Extract(52)。赖氨酸是使用最为频繁的氨基酸。一般来说,赖氨酸占蛋白质氨基酸的6.6%,而甲硫氨酸则仅占1.7%(53)。
生物素化赖氨酸掺入对表达水平和酶活性的影响
赖氨酸残基常见于大多数蛋白质中,通常暴露于亲水区域。生物素化赖氨酸的存在可能会也可能不会影响修饰后蛋白质的功能。在凝胶迁移实验中,TNT®网织红细胞裂解物反应中合成c-Jun,并利用Transcend™ tRNA进行标记,其作用和未标记的c-Jun相同。
评估生物素化赖氨酸的掺入水平
通常,在蛋白质中掺入放射性标记的氨基酸的量是通过掺入标记的百分比表示。这一数值包括假基因产物中放射性的掺入量,例如截短多肽。因此,掺入值的百分比仅能粗略估计合成的全长蛋白质的量,但不能提供任何有关翻译保真度的信息。对于Transcend™ tRNA反应,很难直接确定生物素赖氨酸掺入到翻译的蛋白质中的百分比。另一种估算Transcend™ tRNA反应中翻译效率和保真度的方法是测定SDS-PAGE后可检测产物的最低量。在所有检测情况下,我们使用最少0.5µl的Transcend™ tRNA在50µl翻译反应中,之后取1µl反应产物检测翻译产物(图9)。生物素的掺入量随着Transcend™ tRNA在反应的添加量呈线性增加,最大值约出现在2μl。
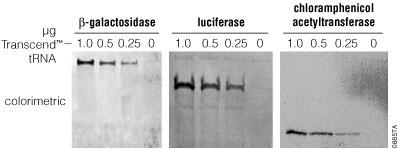
图9.Transcend™ tRNA浓度对检测体外合成的蛋白质的影响。进行转录/翻译偶联反应。在翻译反应中添加一定量的Transcend™ tRNA(相当于2.0、1.0、0.5或0µl),随后在30℃下孵育1小时。1微升反应用于SDS-PAGE中。将分离的蛋白质转移至PVDF膜上(1小时100V)。将膜在TBS + 0.5% Tween®20中封闭15分钟,并利用Streptavidin-AP进行标记(45分钟),利用TBS + 0.5% Tween®20洗涤两次,随后再用TBS洗涤两次,然后使用Western Blue®Substrate孵育2分钟。
捕获生物素化蛋白质
可使用生物素-结合树脂(例如SoftLink™ Soft Release Avidin Resin,目录号:V2011、V2012)从翻译反应中移除生物素化蛋白质。包含多个生物素的新生蛋白质可与SoftLink™ Resin强力结合,不能使用“软释放”在非变性条件洗脱。然而,SoftLink™ Resin可作为免疫沉淀反应的替代品。
翻译产物的比色和化学发光检测
含有生物素的翻译产物可通过以下两种方式进行分析:可利用SDS-PAGE直接分离产物,并将其转移至相应的膜上,然后通过比色或化学发光方法进行检测(图10)。或者,可使用生物素-结合树脂(例如SoftLink™ Resin)从翻译混合物中捕获生物素化蛋白质。该方法可作为蛋白质复合物的免疫沉淀反应的替代方法。
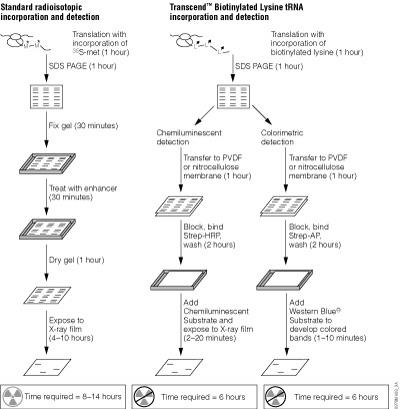
图10.翻译产物的比色和化学发光检测的示意图。
FluoroTect™ GreenLysin vitro Translation Labeling System
FluoroTect™ GreenLysin vitro Translation Labeling System使用带电荷的赖氨酸tRNA分子,在赖氨酸的epsilon(e)氨基酸位点利用荧光基团BODIPY®-FL进行标记(图11)。对于FluoroTect™ System,可选择赖氨酸作为标记的氨基酸,因为赖氨酸是使用最为频繁的氨基酸之一,通常占蛋白质氨基酸的6.6%。可通过使用激光荧光凝胶扫描仪,在2~5分钟内使用“凝胶内”法对标记的蛋白质进行检测。这减少了对蛋白质凝胶操控的任何要求,例如固定/干燥或任何安全、法规或废物处理问题(例如使用放射性标记氨基酸相关问题)。
非同位素“凝胶内”检测也更加方便快捷,避免了传统非同位素系统费时费力的电印迹和检测步骤。有关该系统详细的方案和背景信息,请参看技术手册#TB285。
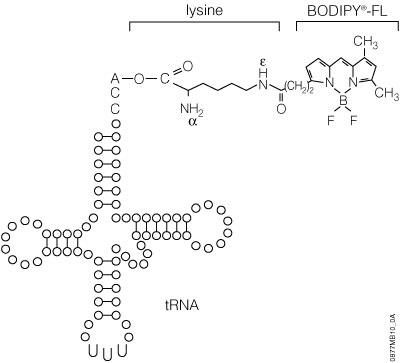
图11.FluoroTect™ GreenLystRNA的结构。
参考文献
1. Zahnd, C. et al. (2007) Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods. 4(3), 269–79.
2. Ginsburg, J. (2009) Decipahering the Genetic Code: A National Historic Chemical Landmark. This can be viewed online at: https://www.acs.org/content/acs/en/education/whatischemistry/landmarks/geneticcode.html#poly-u-experiment.
3. Carlson, E. et al. (2012) Cell-Free Protein Synthesis: Applications Come of Age. Biotechnol. Adv. 30(5), 1185–1194.
4. Chong, S. (2014) Overview of Cell-Free Protein Synthesis: Historic Landmarks, Commercial Systems, and Expanding Applications. Curr. Protoc. Mol. Biol. 108, 16.30.1–11.
5. He, M. (2008) Cell-free protein synthesis: applications in proteomics and biotechnology. New Biotech. 25(2–3), 126–132.
6. Hurst, R. (2011) Innovative Applications for Cell-Free Expression. Promega Corporation Website, accessed June 2019.
7. Pelham, H.R.B. and Jackson, R.J. (1976) An efficient mRNA-dependent translation system from reticulocyte lysates. Eur. J. Biochem. 67, 247–56.
8. Krieg, P. and Melton, D. (1984) Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucl. Acids Res. 12, 7057–70.
9. Rando, R.R. (1996) Chemical biology of protein isoprenylation/methylation. Bichimica Biophysica Acta. 1300, 5–16
10. Han K.-K. and Martinage, A. (1992) Post-translational chemical modification(s) of proteins. Int. J. Biochem. 24, 19–28.
11. Chow, M. et al. (1992) Structure and biological effects of lipid modifications on proteins. Curr. Opin. Cell Biol. 4, 629–36.
12. MacDonald, M.R. et al. (1988) Posttranslational processing of alpha-, beta-, and gamma-preprotachykinins. Cell-free translation and early posttranslational processing events. J. Biol. Chem. 263, 15176–83.
13. Ray, R.B. et al. (1995) Transcriptional regulation of cellular and viral promoters by the hepatitis C virus core protein. Virus Research. 37, 209–20.
14. Bocco, J.L. et al. (1988) Processing of SP1 precursor in a cell-free system from poly(A+) mRNA of human placenta. Mol. Biol. Reports. 13, 45–51.
15. Ezure, T. et al. (2006) Cell-free protein synthesis system prepared from insect cells by freeze-thawing. Biotechnol. Prog. 22, 1570–7.
16. Morita, E.H. et al. (2003) A wheat germ cell-free system is a novel way to screen protein folding and function. Protein Sci. 12, 1216–21.
17. Vinarov, D.A. et al. (2004) Cell-free protein production and labeling protocol for NMR-based structural proteomics. Nature Methods. 12, 149–53.
18. Zubay, G. (1973) In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 7, 267–87.
19. Zubay, G. (1980) The isolation and properties of CAP, the catabolite gene activator. Meth. Enzymol. 65, 856–77.
20. Lesley, S.A. et al. (1991) Use of in vitro protein synthesis from polymerase chain reaction-generated templates to study interaction of Escherichia coli transcription factors with core RNA polymerase and for epitope mapping of monoclonal antibodies. J. Biol. Chem. 266, 2632
21. Pratt, J.M. (1984) In: Transcription and Translation, Hanes, B.D. and Higgins, S.J., eds., IRL Press, Oxford, UK.
22. Studier, F.W. and Moffatt, B.A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes.J. Mol. Biol. 189, 113–30.
23. Collins, J. (1979) Cell-free synthesis of proteins coding for mobilisation functions of ColE1 and transposition functions of Tn3. Gene.6, 29–4
24. Noren, C.J. et al. (1989) A general method for site-specific incorporation of unnatural amino acids into proteins. Science. 244, 182–8.
25. Nand, A. et al. (2012) Emerging technology of in situ cell free expression protein microarrays. Protein & Cell. 3(2), 84–88.
26. He, M. and Taussig, M.J. (2001) Single step generation of protein arrays from DNA by cell-free expression and in situ immobilization (PISA method). Nucleic Acids Res. 29 (15), e73.
27. Manzano-Román, R. and Fuentes, M. (2019) A decade of Nucleic Acid Programmable Protein Arrays (NAPPA) availability: News, actors, progress, prospects and access. J. Proteomics. 198, 27–35.
28. Berrade, L. et al. (2011) Protein Microarrays: Novel Developments and Applications. Pharm. Res. 28(7), 1480–1499.
29. Song, L. et al. (2017) Identification of antibody targets for tuberculosis serology using high-density nucleic acid programmable protein arrays. Mol. Cell. Proteom. 16, 277–289.
30. Wright, C. et al. (2012) Detection of Multiple Autoantibodies in Patients with Ankylosing Spondylitis Using Nucleic Acid Programmable Protein Arrays. Mol. Cell. Proteomics.11(2), M9.00384.
31. Gibson, D. et al. (2012) Circulating and synovial antibody profiling of juvenile arthritis patients by nucleic acid programmable protein arrays. Arthritis Research & Therapy. 14, R77.
32. Anderson, K.S. et al. (2008) Application of protein microarrays for multiplexed detection of antibodies to tumor antigens in breast cancer. J. Proteome Res., 7, 1490–1499.
33. He, M. et al. (2008) Printing protein arrays from DNA arrays. Nature Methods. 5, 175–177.
34. Frey, S. et al. (2007) Synthesis and characterization of a functional intact IgG in a prokaryotic cell-free expression system. Biol. Chem.389, 37–45.
35. Perez, J. et al. (2016) Cell-Free Synthetic Biology: Engineering Beyond the Cell. Cold Spring Harb. Perspect. Biol. 8(12), a023853.
36. Packer, M. and Liu, D. (2015) Methods for the directed evolution of proteins. Nature Reviews Genetics. 16, 379–394.
37. Mie, M. et al. (2008) Selection of mRNA 5’-untranslated region sequence with high translation efficiency through ribosome display. Biochem. Biophys. Res. Commun. 373, 48–52.
38. Weichhart, T. et al. (2003) Functional selection of vaccine candidate peptides from Staphylococcus aureus whole-genome expression libraries in vitro. Infect. Immun. 71, 4633–4641.
39. Lipovsek, D. and Plückthun, A. (2004) In-vitro protein evolution by ribosome display and mRNA display. J. Imm. Methods. 290(1–2), 51–67.
40. Keefe, A.D. and Szostak, J.W. (2001) Functional proteins from a random-sequence library. Nature. 410, 715–718.
41. Seelig, B. and Szostak, J.W. (2007) Selection and evolution of enzymes from a partially randomized non-catalytic scaffold. Nature. 448, 828–831.
42. Xu, L. et al. (2002) Directed evolution of high affinity antibody mimics using mRNA display. Chem. Biol.9, 933–942.
43. Olson, C.A. et al. (2008) mRNA display selection of a high-affinity, modification-specific phospho-IkappaBalpha-binding fibronectin. ACS Chem. Biol. 3(8), 480–5.
44. Miller, O. et al. (2006) Directed evolution by in vitro compartmentalization. Nature Methods. 3, 561–570.
45. Contreras-Llano, L. and Tan, C. (2018) High-throughput screening of biomolecules using cell-free gene expression systems. Synth. Bio. 3(1), ysy012.
46. Xiao, H. et al. (2015) High Throughput Screening and Selection Methods for Directed Enzyme Evolution. Ind. Eng. Chem. Res. 54(16), 4011–4020.
47. Wei, Q. et al. (2005) Toxin detection by a minisaturised in vitro protein expression array. Anal. Chem. 77, 5494–5500.
48. Brandi, L. et al. (2007) Review: methods for identifying compounds that specifically target translation. Methods Enzymol.431, 229–267.
49. Kanter, G. et al. (2007) Cell-free production of scFv fusion proteins: an efficient approach for personalized lymphoma vaccines. Blood. 109, 3393–3399.
50. Hook, B. (2011) Non-Radioactive Detection of Proteins Expressed in Cell-Free Expression Systems. Promega Corporation Website, accessed June 2019.
51. Johnson, A.E. et al. (1976) Nepsilon-acetyllysine transfer ribonucleic acid: A biologically active analogue of aminoacyl transfer ribonucleic acids. Biochem. 15, 569–75.
52. Kurzchalia, T.V. et al. (1988) tRNA-mediated labelling of proteins with biotin. A nonradioactive method for the detection of cell-free translation products. Eur. J. Biochem. 172, 663–8.
53. Dayhoff, M.O. (1978) In: Atlas of Protein Sequence and Structure, Suppl. 2 National Biomedical Research Foundation, Washington, DC.
54. Crowley, K.S. et al. (1993) The signal sequence moves through a ribosomal tunnel into a noncytoplasmic aqueous environment at the ER membrane early in translocation. Cell. 73, 1101–15.




